All content on this site is intended for healthcare professionals only. By acknowledging this message and accessing the information on this website you are confirming that you are a healthcare professional. If you are a patient or carer, please visit Know AML.
The AML Hub website uses a third-party service provided by Google that dynamically translates web content. Translations are machine generated, so may not be an exact or complete translation, and the AML Hub cannot guarantee the accuracy of translated content. The AML Hub and its employees will not be liable for any direct, indirect, or consequential damages (even if foreseeable) resulting from use of the Google Translate feature. For further support with Google Translate, visit Google Translate Help.


The AML Hub is an independent medical education platform, sponsored by Daiichi Sankyo, Johnson & Johnson, Syndax, Thermo Fisher Scientific, Kura Oncology, and AbbVie. Funders are allowed no direct influence on our content. The levels of sponsorship listed are reflective of the amount of funding given. View funders.
Now you can support HCPs in making informed decisions for their patients
Your contribution helps us continuously deliver expertly curated content to HCPs worldwide. You will also have the opportunity to make a content suggestion for consideration and receive updates on the impact contributions are making to our content.
Find out more
Create an account to access:
Bookmark & personalize site content
Receive alerts for new content in your areas of interest
View AML content recommended for you
Association between prior lenalidomide exposure and TP53-mutated therapy-related AML
Do you know... Prior exposure to which of the following treatments is positively associated with TP53 mutations?
Therapy-related myeloid neoplasms (t-MN) are secondary malignancies, such as acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS).1 t-MN arise as a result of prior exposure to chemotherapy and/or radiation therapies (CRTs) for a primary condition. Generally, t-MN develop 3–7 years after exposure to CRTs and confer an adverse prognosis. Recent data suggest that t-MN may arise due to CRTs inducing clonal selection of preexisting mutant hematopoietic stem and progenitor cells (HSPCs).1
The thalidomide analogs lenalidomide and pomalidomide facilitate the degradation of Ikaros transcription factors IKZF1 and IKZF3.1 Lenalidomide has also been shown to promote the degradation of the protein kinase CK1α.1 Sperling et al.1 recently published an article in Blood suggesting that prior exposure to lenalidomide promotes the development of TP53-mutated t-MN, and we are pleased to summarize the key findings below.
Study design
Patients and mutational profiling
A retrospective analysis was performed of data from 416 patients diagnosed with t-MN (based on the 2016 World Health Organization classification) at MD Anderson Cancer Center, US, between 2008 and 2019. A comparison was made with data from 1,021 patients with de novo MN who were diagnosed in the same time period.
Next-generation sequencing was used to detect somatic mutations in bone marrow (BM) and peripheral blood samples using a 300-gene panel (n = 156).
Mouse model experiments
Trp53 mutations and resistance to thalidomide analogs
Immortalized mouse HSPC cell lines with an estrogen-inducible Hoxb8 transgene were generated using CrbnI391V knock-in mice.
Hoxb8 CrbnI391V;Rosa26-Cas9 cells were engineered to carry recurrent clonal hematopoiesis mutations using the CRISPR (clustered regularly interspaced short palindromic repeats)-Cas9 system and treated with lenalidomide and pomalidomide.
Selective advantage of HSPCs in the presence of thalidomide analogs
Live, lineagelo, Sca1+, c-Kit+ (LSK) cells from CrbnI391V;Rosa26-Cas9 were virally transduced with concentrated lentiviruses encoding single-guide RNAs targeting Dnmt3a, Tet2, Asxl1, Trp53, Ppm1d, and Ezh2 genes, a tag-red fluorescent protein, and two control single-guide RNAs in parallel. Mutant cells were transplanted into lethally irradiated wild-type (WT) recipient mice and validated through next-generation sequencing and flow cytometry (Figure 1).
c-Kit+ cells were transplanted with donor marrow consisting of 20% CD45.2;Trp53−/−;CrbnI391V cells mixed with 80% CD45.1;CrbnI391V WT cells. Mice were then treated with dimethyl sulfoxide, lenalidomide 50 mg/kg, or pomalidomide 20 mg/kg.
Figure 1. Schematic of mouse model experiments to determine the selective advantage of HSPCs in the presence of thalidomide analogs
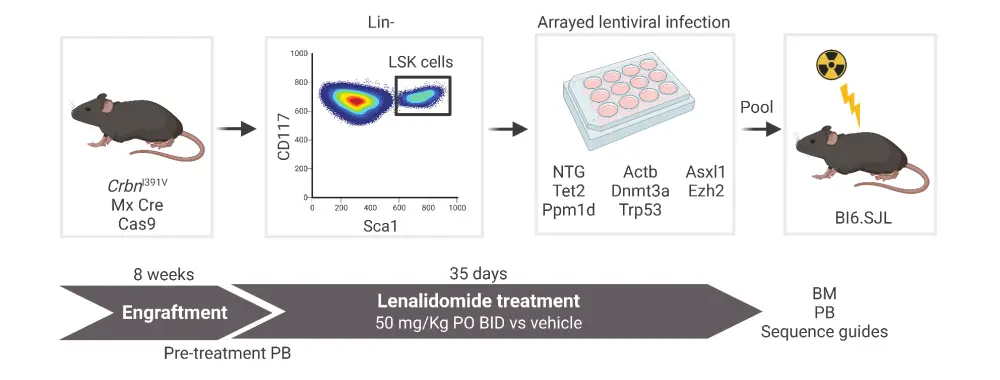
BID, twice a day; BM, bone marrow; HSPC, hematopoietic stem and progenitor cell; LSK, Live, lineagelo, Sca1+, c-Kit+; PB, peripheral blood; PO, taken orally.
*Adapted from Sperling, et al.1 Created with BioRender.com.
Differential toxicity of thalidomide analogs in HSPCs
Competitive BM transplant experiments were carried out with Csnk1a1 heterozygous knock-out mouse cells and WT cells in the Crbn1391V background. Transplanted mice were treated with dimethyl sulfoxide , lenalidomide 50 mg/kg, pomalidomide 20 mg/kg, or iberdomide 20 mg/kg.
Results
Patient characteristics
Of the 416 patients diagnosed with t-MN, a total of:
- 40% were diagnosed with therapy-related AML and 60% with therapy-related MDS
- 63% had a primary diagnosis of solid tumors and 37% had non-myeloid hematologic cancers
- 45% received prior treatment with chemotherapy alone, 17% with radiotherapy alone, and 39% received both
- 17% underwent hematopoietic stem cell transplantation
Statistically relevant patient characteristics are listed in Table 1.
Table 1. Characteristics of patients diagnosed with t-MN*
|
PLT, platelet; t-AML, therapy-related acute myeloid leukemia; t-MDS, therapy-related myelodysplastic syndromes; WBC, white blood cell. |
||||
|
Characteristic, % (unless otherwise specified) |
All patients (N = 416) |
t-AML (n = 167) |
t-MDS (n = 249) |
p value |
|---|---|---|---|---|
|
Median age (range), years |
68 (17–91) |
65 (17–89) |
69 (22–91) |
0.002 |
|
Median latency† (range), years |
6.0 (0.1–40) |
5.0 (0.1–40) |
6.4 (0.3–45) |
0.028 |
|
Median WBC (range), K/μL |
3.2 (0.1–267) |
3.8 (0.1–267) |
3 (0.2–85.7) |
<0.001 |
|
Median PLT (range), K/μL |
58 (3–895) |
41 (3–389) |
65 (6–895) |
0.002 |
|
Active primary malignancy |
15 |
7 |
21 |
<0.001 |
|
Cytogenetics‡ |
|
|
|
|
|
Del 5q/-5§ |
32 |
25 |
37 |
0.013 |
|
Del 7q/-7 |
34 |
20 |
43 |
<0.001 |
|
Inv 16/t (16;16) |
1 |
4 |
0 |
0.003 |
|
11q23 |
7 |
16 |
1 |
<0.001 |
|
t (15;17) |
1 |
2 |
0 |
0.036 |
|
t (8;21) |
1 |
2 |
0 |
0.036 |
Mutational profiling
At least one gene mutation was detected in 85% of patients with t-MN. The most common gene mutations detected were TP53 (37%), PPM1D (19%), TET2 (16%), DNMT3A (15%), RUNX1 (13%), ASXL1 (13%), and SRSF2 (10%). Comparison with the de novo AML and MDS cohort (N = 1,021) found that:
- TP53 and PPM1D mutations were significantly associated with t-MN
- STAG2 and ASXL1 mutations were more likely to occur in de novo AML/MDS
- NPM1, IDH1/2, FLT3, CEBPA, and NRAS mutations occurred more frequently in de novo AML
- TET2, PHF6, SRSF2, SF3B1, and U2AF1 mutations were more common in de novo MDS
Analysis based on treatment
Complex karyotype was associated with prior exposure to platinum agents (odds ratio [OR], 1.88; 95% confidence interval [CI], 1.23–2.89; false discovery rate [FDR] = 0.052).
Abnormalities in chromosome 7 were associated with prior exposure to alkylating agents (OR, 1.64; 95% CI, 1.08–2.49; FDR = 0.057) and platinum agents (OR, 1.65; 95% CI, 1.06–2.57; FDR = 0.057).
Patients who received radiation therapy were more likely to harbor NPM1 mutations, splicing gene mutations, and normal karyotype.
Mutations in TP53 were associated with treatment with proteasome inhibitors (OR, 3.06; 95% CI, 1.52–6.15; FDR = 0.025), and thalidomide analogs (OR, 2.62; 95% CI, 1.36–5.05; FDR = 0.035). Multivariate logistic regression analysis also found TP53 mutations were more common in patients treated with thalidomide analogs (OR, 3.14; 95% CI, 1.60–6.18; p = 0.009) or vinca alkaloids (OR, 1.76; 95% CI, 1.05–2.93; p = 0.031), and less common in patients treated with topoisomerase inhibitors (OR, 1.76; 95% CI, 1.05–2.93; p = 0.031).
In patients treated with lenalidomide, TP53 mutations were associated with a longer duration of exposure.
Mouse model experiments
In vivo and in vitro mouse models demonstrated that lenalidomide, but not pomalidomide, conferred a selective advantage to Trp53-mutant HSPCs.
- Multiplexed CRISPR-Cas9 experiments showed that Trp53 mutations conferred a selective advantage to HSPCs in the presence of lenalidomide, whereas mutations in Dnmt3a, Tet2, Asxl1, Trp53, Ppm1d, and Ezh2 genes did not.
- In long-term in vitro competition assays, mutated Trp53 cells demonstrated a strong resistance to lenalidomide compared with control cells, whereas pomalidomide only had a mild proliferation advantage even at high doses.
- In competitive BM transplant experiments with Csnk1a1−/+ cells, differential HSPC toxicity occurred mainly due to lenalidomide-specific CK1α degradation.
Key findings
- Mutational profiling revealed that genes enriched in patients with t-MN differed from patients with de novo AML/MDS, with TP53 and PPM1D mutations occurring more frequently in patients with t-MN, suggesting selective pressure from prior therapies may drive t-MN.
- A positive association was found between exposure to thalidomide analogs and the presence of TP53 mutations.
- Mouse model experiments demonstrated that lenalidomide, but not pomalidomide, exerted selective pressure on Tp53-mutant leukemic cells and HSPCs, but was not associated with the proliferation of cells with other common clonal hematopoietic mutations. Moreover, lenalidomide was linked to Tp53 mutant selection, likely due to the degradation of CK1α.
- Further experiments with pomalidomide are necessary, as pomalidomide exposure normally occurs after prior exposure to lenalidomide, making it challenging to extract the independent effect of pomalidomide alone.
This study also highlights the importance of genetic screening prior to initiation of treatment, to identify potential mutations that may exert selective pressure and to improve risk stratification.
References
Please indicate your level of agreement with the following statements:
The content was clear and easy to understand
The content addressed the learning objectives
The content was relevant to my practice
I will change my clinical practice as a result of this content